A patient with painful mouth ulcers and itchy skin lesions
Test your diagnostic skills in our regular dermatology quiz. What is the cause of these worsening oral erosions and newer cutaneous lesions?
Case presentation
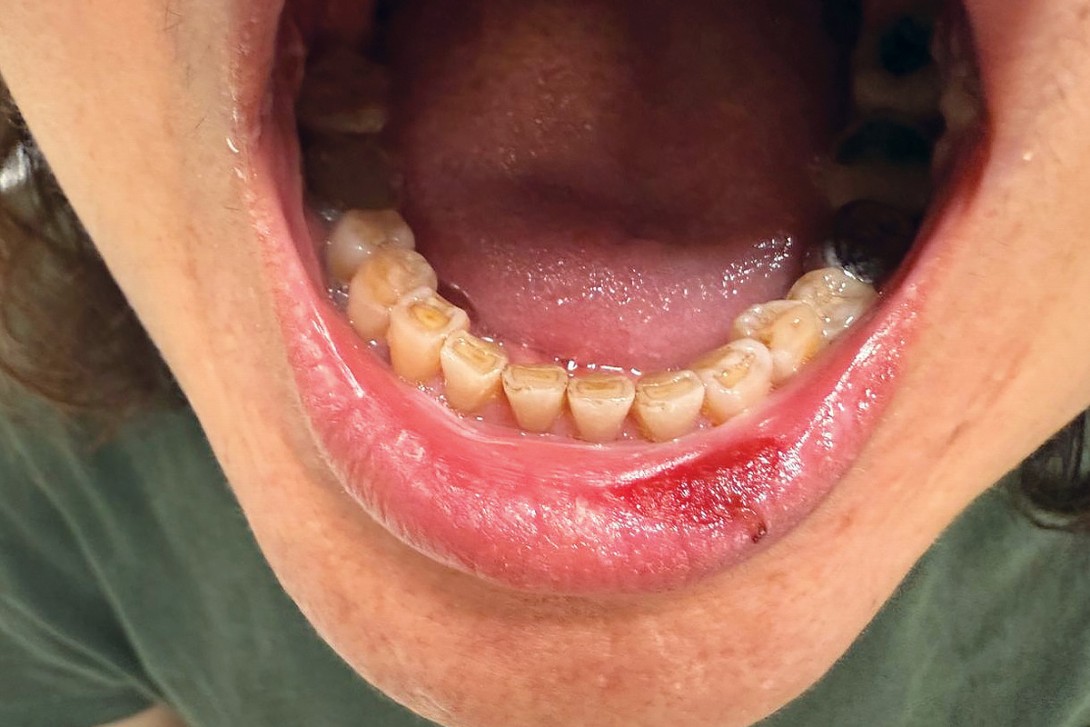
A 52-year-old woman presents with painful erosions affecting her lips and oral mucosa (Figures 1a and b). The lesions have been present for several months and have been progressively worsening over the preceding few weeks, causing severe pain that is limiting her ability to swallow, eat and drink. She has also developed a scaly eruption on her scalp that is intensely pruritic and new similar lesions on her trunk (Figure 2).


The patient has no significant past medical history. She takes no regular medications and reports no drug allergies.
On examination, superficial erosions are observed involving the patient’s vermilion lip, buccal mucosa and the hard and soft palate. Desquamative gingivitis is also noted. Her scalp shows hyperkeratosis with prominent scale. There are scattered erythematous macules and superficial erosions on the trunk. There is no involvement of the genital mucosa.
Differential diagnoses
Conditions to consider among the differential diagnoses include the following.
Erosive oral lichen planus
Oral lichen planus is a chronic inflammatory mucosal disorder that commonly affects the buccal mucosa, tongue and gingivae.1 A subtype of lichen planus, it is often seen in middle-aged adults. Patients may present with white striae, erythema or painful erosions, which can significantly impair oral intake. Reticular white streaks on the buccal mucosa are classic but may be subtle or absent in predominantly erosive disease.2 Oral involvement may coexist with genital disease.3 Biopsy and histopathology typically shows a lichenoid interface dermatitis and may be aided by direct immunofluorescence (DIF) microscopy in select cases.2
For the case patient, the possibility of oral lichen planus was considered, but the combination of extensive, painful oral erosions and the more recent appearance of erosive cutaneous lesions raised concern for an alternative diagnosis. Histopathology with immunofluorescence testing was important to distinguish between these possibilities.
Herpes simplex virus stomatitis
Primary oral herpes simplex virus (HSV) infection can cause widespread, painful vesiculoulcerative lesions affecting the lips, gingivae, buccal mucosa and palate. Systemic symptoms and cervical lymphadenopathy may also occur. HSV-1 is the type most commonly associated with oral disease.4
Primary oral HSV infection is usually acute and recurrent disease is often localised (e.g. herpes labialis), but more extensive mucosal involvement can occur in immunocompromised patients. Diagnosis is often clinical but may be confirmed by laboratory testing (e.g. polymerase chain reaction [PCR] assay) when the presentation is atypical or severe.4
For the case patient, the chronicity of the oral lesions (over a period of months) and the development of new cutaneous erosions made HSV stomatitis alone a less likely diagnosis. However, HSV infection can complicate other mucosal disorders and should be considered if lesions are unusually acute, clustered or necrotic.
Aphthous ulceration
Aphthous ulceration is the most common ulcerative condition of the oral mucosa. Patients typically present with shallow, round ulcers, often on nonkeratinised mucosa (e.g. on the inner side of the lips and cheeks or under the tongue).5 The lesions are painful but usually self-limiting and heal without scarring. Although aphthous ulcers may be associated with systemic disease (e.g. inflammatory bowel disease, nutritional deficiencies, viral infections), they do not usually cause widespread erosive disease of keratinised mucosa (such as the hard palate) or an accompanying blistering or erosive cutaneous eruption.5
This was not the correct diagnosis for the case patient. The distribution and severity of her mucosal involvement and the presence of new trunk and scalp lesions were more suggestive of a broader mucocutaneous pathological process.
Stevens-Johnson syndrome and toxic epidermal necrolysis
Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) are severe mucocutaneous reactions, most often drug-induced, characterised by painful mucosal erosions with an acute onset of widespread dusky or targetoid lesions and epidermal detachment.6 Patients are typically systemically unwell, with prodromal fever and malaise, and disease progression is measured in days. Early recognition and urgent supportive management are essential because of the high morbidity and mortality.6 Histopathology is crucial when clinical uncertainty exists.
For the case patient, the prominent mucosal erosions and trunk involvement warranted consideration of SJS/TEN on presentation. However, the prolonged time course (a period of months), absence of a clear new drug exposure and the lack of typical widespread purpuric macules and erosions were not consistent with SJS/TEN.
Pemphigus vulgaris
This is the correct diagnosis. Pemphigus vulgaris most commonly presents in adults aged 30 to 60 years, although it can occur at any age including, uncommonly, in children.7 It is a rare autoimmune blistering disease characterised by painful mucosal erosions and fragile, flaccid cutaneous blisters that rupture easily to form erosions.8 Pemphigus vulgaris is caused by pathogenic IgG autoantibodies directed against desmosomal proteins (classically desmoglein 3, often with desmoglein 1), leading to loss of keratinocyte adhesion (acantholysis) and formation of the intraepidermal blisters.9 Oral mucosal involvement is common and may precede cutaneous disease by weeks or months, often presenting as persistent, painful erosions that impair ability to eat and drink.
For the case patient, the history of severe painful oral erosions over several months followed by the development of newer erosions on the scalp and trunk was characteristic of pemphigus vulgaris.
Diagnosis
The diagnosis of pemphigus vulgaris requires compatible clinical features plus supportive histopathological and immunopathological findings. The current expert recommendations emphasise that diagnosis requires both of the following.10
- Histopathology findings that are consistent with pemphigus. A lesional biopsy specimen for routine histology typically shows suprabasal acantholysis with a characteristic ‘tombstone’ appearance of basal keratinocytes attached to the basement membrane.
- Evidence of autoantibodies against epithelial cell surface antigens (by perilesional biopsy and DIF microscopy and/or by serological detection). For autoimmune bullous disease, the optimal biopsy site for DIF microscopy is normal-appearing skin close to a blister; a positive result for pemphigus vulgaris is demonstrated by intercellular IgG (± complement C3) deposition in an intercellular (‘chicken-wire’) pattern in the specimen. Serological evidence of circulating intercellular antibodies may be demonstrated by indirect immunofluorescence or ELISA measurement of anti-desmoglein 1 and 3 titres (which may also be helpful to monitor disease activity).10
The management of pemphigus vulgaris often requires prolonged systemic immunosuppression, so baseline screening for latent infections that may reactivate should also be arranged, alongside a vaccination and immunity review. This includes testing for hepatitis B, hepatitis C, HIV, interferon- gamma release assay (e.g. Quantiferon-TB Gold) and strongyloides.11
Management
The goals of management for pemphigus vulgaris are to achieve rapid control of disease activity (stop new blister formation and heal erosions), prevent complications (infection, fluid or protein loss, malnutrition) and minimise treatment-related adverse effects. Management is often complex, and referral to a dermatologist or oral medicine specialist is recommended.8,12
Management includes a combination of systemic and topical immunosuppressants. Systemic corticosteroids (prednisone, prednisolone) have been the mainstay of therapy. Corticosteroid-sparing immunosuppressants (such as azathioprine, mycophenolate mofetil or ciclosporin) are commonly used to reduce cumulative corticosteroid exposure.10
The monoclonal treatment rituximab (anti-CD20 therapy) has become a key treatment for moderate-to-severe disease, with randomised trial data support improved sustained remission outcomes and corticosteroid-sparing effects when used early in treatment.8,13 In Australia, rituximab use for pemphigus vulgaris is off-label and requires specialist oversight. Less commonly, cyclophosphamide is used as a steroid-sparing rescue option for severe, refractory disease when other agents are unsuitable, but its role is limited by toxicity and infection risk.
Topical therapy is an important adjunct. Potent topical corticosteroids may be used on cutaneous lesions, and topical corticosteroid mouth rinses can help achieve symptomatic control of oral disease. Secondary infection (including candidiasis) should be treated. Supportive care, including analgesia and nutritional support, is essential when oral intake is compromised.
Before escalating immunosuppression, pre-treatment screening and a plan for ongoing monitoring are essential. The growing use of immunomodulatory drugs and need for screening is highlighted in Therapeutic Guidelines, with recommendations for monitoring and strategies to manage intercurrent infection in patients receiving these therapies.11,14 Particular attention should be paid to the risk of hepatitis B reactivation with rituximab. Hepatitis B serology (including core antibody) should be checked prior to treatment, and antiviral prophylaxis should be discussed for patients who have serological evidence of past or chronic infection.15
Outcome
Investigations were arranged for the case patient to evaluate for inflammatory and blistering disorders. Lesional and perilesional biopsy specimens were obtained from an active lesion on the trunk and serological testing was requested. Histopathology for the truncal lesion demonstrated suprabasal acantholysis with basal keratinocytes attached to the basement membrane (‘tombstone’ appearance). DIF microscopy for the perilesional specimen showed an intercellular ‘chicken‑wire’ pattern of IgG and C3 deposition (and were negative for IgA and IgM). Together with clinical findings, these results confirmed an autoimmune intraepidermal blistering disorder consistent with pemphigus vulgaris. The serology results supported the diagnosis, with circulating intercellular antibodies (titre 1:80) and positive anti‑desmoglein 1 and 3 antibodies. Due to her swallowing difficulties, a gastroscopy was undertaken and to exclude oesophageal involvement, which showed superficial lesions in the buccal mucosa and one or two pemphigoid lesions in the crico-oesophagus. The rest of the oesophagus was unremarkable.
Initial management comprised systemic corticosteroid therapy (prednisone 25 mg daily), with early introduction of mycophenolate mofetil (750 mg twice daily) as a corticosteroid-sparing agent. In addition, topical corticosteroids were commenced: triamcinolone acetonide 0.1% intraorally and betamethasone dipropionate 0.05% for scalp and cutaneous disease. Given the severity of oral involvement, a dexamethasone mouthwash was also used for symptom control and an antifungal agent was added to reduce the risk of secondary candidiasis. The cutaneous eruption improved but the oral disease remained the major driver of morbidity, causing ongoing pain and reduced oral intake, and there was subsequent involvement at other mucosal sites.
Pre‑treatment screening for escalation of immunosuppression was negative for active infection, but hepatitis B serology indicated past infection (core antibody positive). Under the advice and management of a gastroenterologist, antiviral risk mitigation was instituted (daily entecavir). Rituximab therapy was commenced when hepatitis B status was appropriately addressed. Supportive measures to optimise nutrition, pain control and psychological wellbeing were implemented concurrently.
At a three-month follow-up visit, substantial improvement was observed, with clearance of the scalp and other cutaneous lesions and residual but improving oral lesions. MT
COMPETING INTERESTS: None.
References
1. Nogueira PA, Carneiro S, Ramos-e-Silva M. Oral lichen planus: an update on its pathogenesis. Int J Dermatol 2015; 54: 1005-1010.
2. Mirowski GW, Culton DA. Oral lichen planus: pathogenesis, clinical features, and diagnosis. In: Connor R (ed). UpToDate. Waltham (MA): Wolters Kluwer, 2025.
3. Krupaa RJ, Sankari SL, Masthan KM, Rajesh E. Oral lichen planus: an overview. J Pharm Bioallied Sci 2015; 7 (Suppl 1): S158-161.
4. Oakley A. Herpes simplex. DermNet NZ; 2015. Available online at: https://dermnetnz.org/topics/herpes-simplex (accessed March 2026).
5. Akintoye SO, Greenberg MS. Recurrent aphthous stomatitis. Dent Clin North Am 2014; 58: 281-297.
6. Lee HY. Stevens-Johnson syndrome and toxic epidermal necrolysis: management, prognosis, and long-term sequelae. In: Connor RF (ed). UpToDate. Waltham, MA: Wolters Kluwer, 2025.
7. Ngan V, Oakley A. Pemphigus vulgaris. DermNet NZ; 2022 [updated by Shakeel M]. Available online at: https://dermnetnz.org/topics/pemphigus-vulgaris (accessed March 2026).
8. Blistering skin conditions: Pemphigus vulgaris: oral features. In: Therapeutic Guidelines: oral and dental. Melbourne: TG, 2025. Available online at: https://www.tg.org.au/ (accessed March 2026).
9. Black M, Mignogna M, Scully C. Pemphigus vulgaris. Oral Dis 2005; 11: 119-130.
10. Murrell DF, Peña S, Joly P, et al. Diagnosis and management of pemphigus: recommendations of an international panel of experts. J Am Acad Dermatol 2020; 82: 575-585.571.e1.
11. Ashok A, Macphail AI, Giles ML, Gardiner BJ. Preventing infections in immunosuppressed patients. Aust Presc 2026; 49: 22-29.
12. Blistering skin conditions. In: Therapeutic Guidelines: dermatology. Melbourne: TG, 2022. Available online at: https://www.tg.org.au/ (accessed March 2026).
13. Joly P, Maho-Vaillant M, Prost-Squarcioni C, et al. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet 2017; 389: 2031-2040.
14. Principles of immunomodulatory drug use. In: Therapeutic Guidelines: rheumatology. Melbourne: TG; 2024. Available online at: https://www.tg.org.au/ (accessed March 2026).
15. Cholongitas E, Haidich AB, Apostolidou-Kiouti F, Chalevas P, Papatheodoridis GV. Hepatitis B virus reactivation in HBsAg-negative, anti-HBc-positive patients receiving immunosuppressive therapy: a systematic review. Ann Gastroenterol 2018; 31: 480-490.
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.